Par Anatole von Lilienfeld
L’introduction de l’IA dans le mélange des simulations informatiques basées sur la physique a entraîné des changements fascinants dans le monde de la chimie et des sciences des matériaux. Au lieu de passer des heures à fouiner dans le noir à étudier un composé à la fois, nous avons maintenant des gadgets d’apprentissage automatique qui trient des montagnes de composés virtuels, fournissent des réponses fiables sur de nouveaux composés chimiques ou de nouvelles façons d’ajuster des matériaux existants pour des tâches spécifiques.
Ce changement s’apparente à l’introduction de calculatrices remplaçant les calculs à main. Cependant, dans ce cas, les calculatrices nous offrent un raccourci formidable pour comprendre et contrôler. Il pourrait bientôt devenir courant de fabriquer à la demande et en temps réel les matériaux mêmes dont notre monde est fait, ce qui nous permettra de relever de nombreux défis mondiaux grâce à des matériaux et molécules améliorés. Imaginez comment cela nous permettra d’accélérer la découverte de médicaments et la médecine personnalisée, ou de réduire notre empreinte carbone en créant de nouvelles technologies de batteries.
Je trouve remarquablement beau de mêler ces anciennes et nouvelles façons de penser, une approche illustrée par les trois publications suivantes de mon laboratoire.
La théorie de la fonction de la densité à l’ère de l’IA
La mécanique quantique, en particulier une approche appelée théorie de la fonction de la densité (DFT), est un outil puissant pour comprendre les produits chimiques et les matériaux ainsi que pour prédire leurs propriétés et leur comportement. Cette méthode est prisée pour sa précision, sa polyvalence et son universalité dans les simulations informatiques et est largement applicable dans le tableau périodique de Mendeleïev. Nous avons récemment fait beaucoup de progrès dans l’utilisation de modèles d’apprentissage automatique (ML) entraînés sur des données synthétiques obtenues à partir de simulations DFT exigeantes en calcul. Déjà en 2011, nous pouvions démontrer qu’après entraînement, ces modèles d’apprentissage automatique pouvaient fournir des prédictions instantanées pour de nouveaux composés dotés de la qualité et de la polyvalence DFT. Des avancées plus récentes, telles que l’IA générative et les grands modèles de langage, ont eu un impact majeur supplémentaire et préparent le terrain pour des logiciels qui offrent un grand potentiel pour planifier des validations expérimentales réussies dans des laboratoires qui pourraient essentiellement s’exécuter d’eux-mêmes.
Dans l’article « Le rôle central de la théorie de la fonction de la densité à l’ère de l’IA » publié dans Science, nous discutons de la manière dont cela permet un avenir où les expériences dirigées par des robots deviendront aussi fondamentales pour la science que l’apprentissage automatique, les simulations informatiques, les théories traditionnelles et les expériences dirigées par l’humain.
Apprentissage automatique quantique
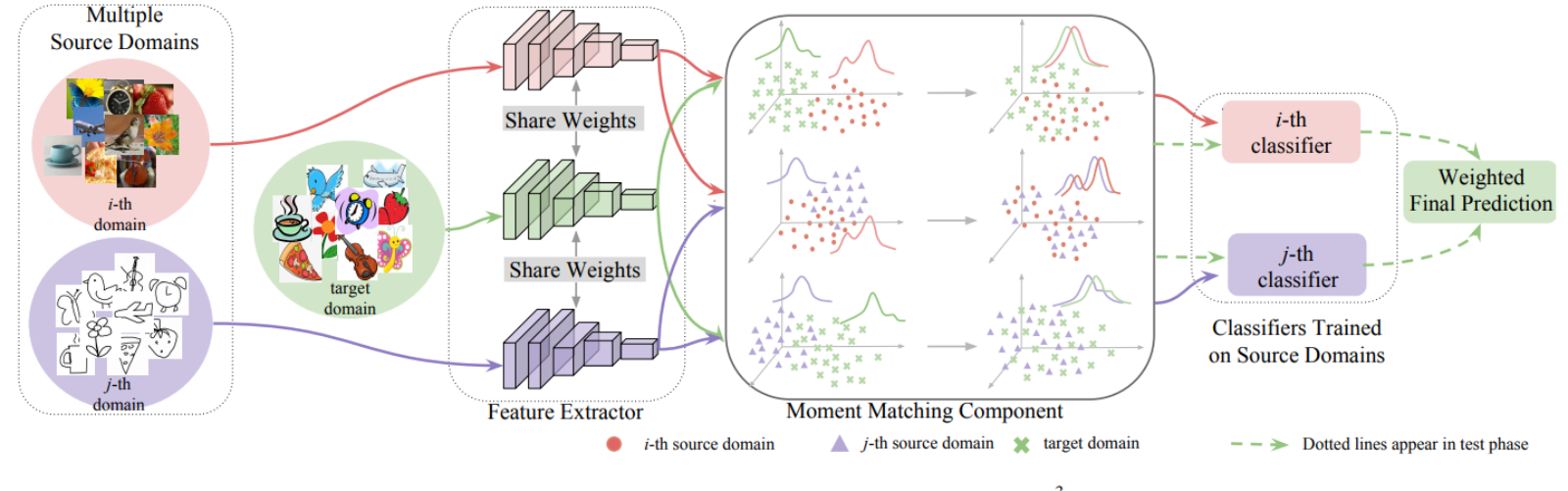
Dans le domaine de nos modèles d’apprentissage automatique basés sur la mécanique quantique, la façon dont nous représentons ou « cartographions » les systèmes chimiques est cruciale, car elle affecte directement les besoins en données d’entraînement nécessaires pour atteindre la puissance prédictive souhaitée. Les méthodes de représentation les plus prédictives, qui s’en sortent avec des tailles minimales d’ensembles de données d’entraînement, tendent cependant à être lourdes en calcul, rendant l’étape d’entraînement lente et exigeante en termes de besoins matériels et d’empreinte carbone.
Depuis que j’ai déménagé mon laboratoire à Toronto l’été dernier, le chercheur postdoctoral Vector Stefan Heinen, l’étudiant diplômé Vector Danish Khan et moi travaillons sur une nouvelle caractérisation des composés chimiques basée sur la physique, si compacte que le coût d’entraînement des modèles ML peut être réduit de plusieurs ordres de grandeur. Dans notre article « Apprentissage quantique basé sur le noyau à rythme record : Fonctionnales de distribution à plusieurs corps comme représentations compactes », publié dans The Journal of Chemical Physics, nous proposons une façon améliorée de représenter les systèmes chimiques qui est non seulement ultra-compacte, mais aussi invariante par rapport à la taille du système. Elle est basée sur ce qu’on appelle les fonctionnelles de distribution atomiques gaussiennes à plusieurs corps (MBDF).
Lorsque nous avons testé le MBDF sur des ensembles de données de référence composés de molécules organiques, sa performance égalait voire rivalisait avec les meilleures méthodes actuelles pour diverses propriétés quantiques. Nos résultats suggèrent que l’approche basée sur le MBDF peut naviguer efficacement l’équilibre entre le coût de l’échantillonnage et de l’entraînement tout en maintenant une grande précision, ce qui en fait le choix privilégié pour certains paramètres de données d’entraînement disponibles et du matériel de calcul. En générant des prédictions chimiquement précises des propriétés quantiques de composés hors échantillon invisibles, il atteint des taux d’échantillonnage dans l’espace des composés chimiques qui reviennent à tamiser environ 48 molécules par seconde à l’aide d’un seul noyau de calcul. Bien que les humains soient généralement incapables de fournir des estimations quantitatives des propriétés quantiques, résoudre numériquement les équations correspondantes pour une seule molécule prendrait facilement des milliers de secondes de façon conventionnelle sans apprentissage automatique.
Accélérer la recherche scientifique par l’automatisation
Cet été, Siwoo Lee, étudiant de premier cycle au département de chimie de l’Université de Toronto, Heinen, Khan et moi avons développé un flux de travail autonome qui combine un réseau de neurones convolutionnel avec un grand modèle de langage pour extraire des tables de données spécifiques d’articles scientifiques. Nous avons testé cette approche pour 592 molécules organiques étudiées dans 74 articles différents publiés entre 1957 et 2014. Ces articles rapportaient des mesures expérimentales sur une propriété cruciale pour la recherche électrochimique, le potentiel d’oxydation, avec des valeurs allant de -0,75 à 3,58 V et le signe indiquant si la molécule préférait attirer ou libérer un électron.
Après avoir sélectionné les données pour validation et pour tenir compte des conditions expérimentales différentes, nous l’avons utilisée pour entraîner des modèles d’apprentissage automatique supplémentaires capables de prédire les potentiels d’oxydation avec une marge d’erreur proche de ce que l’on attendrait d’erreurs expérimentales régulières (environ +/- 0,2 V). Si plusieurs études donnaient des résultats pour la même molécule, notre modèle d’IA pourrait décider quelle valeur est la plus probablement la bonne. Nous avons ensuite utilisé nos modèles pour prédire le potentiel d’oxydation de plus de 100 000 molécules organiques, trouvant des valeurs comprises entre 0,21 et 3,46 V. Notre analyse a montré que certaines caractéristiques moléculaires, comme l’aliphatique, pouvaient augmenter le potentiel d’oxydation d’une moyenne de 1,5 à 2,0 V, tandis que la présence d’un plus grand nombre d’atomes le diminuait généralement. Il est important de noter que notre flux de travail démontre comment la mise en chaîne de plusieurs modèles d’IA permet un flux automatique qui réduira considérablement le travail manuel que les scientifiques devraient normalement faire pour obtenir des estimations des propriétés computationnelles de nouveaux composés basées sur des modèles d’IA entraînés sur des données littéraires.
Cette nouvelle œuvre passionnante a été soumise pour publication et est actuellement disponible en ligne en prépublication. Les applications futures de cette ligne de recherche pourraient bien contribuer à la révolution des efforts de recherche en matériaux expérimentaux et en chimie grâce à des agents IA autonomes qui enquêtent sur des questions et des problèmes inédits grâce à l’utilisation de laboratoires autonomes.
On est sur le point de faire des choses vraiment intéressantes; Nous pourrions bientôt découvrir de nouveaux médicaments, fabriquer de meilleures batteries, produire de l’électronique organique améliorée, concocter des moyens plus propres de déclencher des réactions chimiques avec des catalyseurs sur mesure, et peut-être, juste peut-être, tomber sur ces insaisissables supraconducteurs à température ambiante.